Software to analyze your DNA sequence data with unparalleled clarity.
State of the art analysis for multiple genomes
The best software tool available for viewing, searching, and analyzing the results from many genomes side-by-side
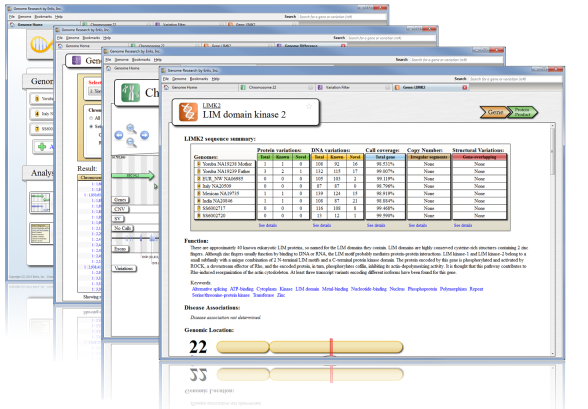
Comprehensive variation annotation
Our proprietary annotation pipeline is combined with a curated set of the best publicly available information. All annotation data is included and formatted - no external downloads are required.

Combine different types of DNA sequence data and analyze them together
Whole genome - Exome - Targeted gene panels - SNP chip
Single nucleotide variants (SNV, SNP) - Insertions - Deletions - Structural variations - Copy number variation
Data from Illumina - Life Technologies - Complete Genomics and more
Data formats: VCF, Complete Genomics, GVF, DNANexus, Affymetrix, CASAVA, and more

Easy File Handling
All relevant variation data on a single genome is assembled into an easy to handle .genome file.

 Analysis Tools
Analysis Tools
Crazy fast multi-genome variation filter. Highly optimized variation searching with a point-and-click interface. Filter millions of variations down to a handful of relevant variations in a matter of seconds.
Search tens, hundreds, or thousands of genomes at the same time. Tested to >10,000 genomes in a single query.
Example Query: Find all the protein-coding variations with a global allele frequency of <1%, found in genes involved in cholesterol metabolism, that are shared by a mother and daughter, not found in the father, and not found in 52 additional control genomes. (Whole genome data)
Results: 6 variations found in 35 seconds

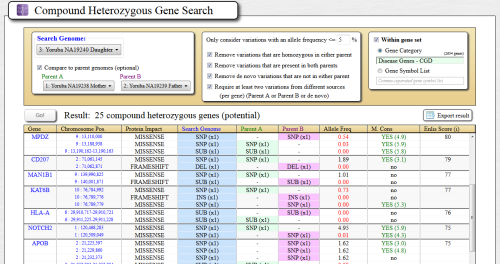
Compare a family trio, or analyze individual genomes for genes that are potentially compound heterozygous. (Different variations are inherited within the same gene)
Genes are scored for impact severity using our highly-tuned algorithm that has shown to accurately recapitulate compound heterozygous genes implicated in many rare disease publications.
Search for keywords that correspond to a list of known phenotypes (Over 6000 diseases and traits). Then, variations that are associated with that phenotype are available to be immediately checked in the currently loaded genomes.
See not only if a genome has variations that are implicated in the phenotype, but also assess how well the genome was called at the positions that are implicated in that phenotype.
Example: Out of 211 known pathogenic variations for Cystic Fibrosis, this individual matches 1 pathogenic variation, 2 of the positions are not called in the sequence data, and the individual matches reference at 208 positions.

Quickly detect genome-wide homozygous regions and compare them to other genomes with a visual chromosome map and results table.
The tool will find regions of the genome with "runs" of consecutive homozygous variants. These regions are often associated with recessive disease or traits, and in cancer samples can indicate loss of heterozygosity.
Preset settings are optimized for exome, or whole genome data. Exome analysis is complete within seconds, and whole genome analysis typically takes about 1 minute.

Fast and responsive chromosome map allows the visualization of multiple types of variation data, in the context of genes and other chromosome features.
Example: A structural variation found in an intron of the gene PQLC2 correlates perfectly with a red region of uncalled sequence, indicating a homozygous deletion, and giving confidence that this is a real event.

Useful for finding known important variations within genomes. Assess at a glance, all the currently loaded genomes for variations found at specific positions. Also determines if the position was successfully called or was not sequenced, and if there are any overlapping copy number or structural variations.
Define custom categories to find positions relevant to your research.
Example: Assess a three-generation 17 member family pedigree at 72 positions that are known to be involved in cystic fibrosis.
Results: Found 1 variation implicated in cystic fibrosis that is inherited in a mendelian fashion. 2 positions were not successfully sequenced. The remaining positions were sequenced and matched the reference genome. Determined in 13 seconds

View a gene category and get a broad overview of the types of variation found the loaded genomes - numbers of protein variation, rare protein variation, rare DNA variation, structural variations and copy number variations.
Over 19,000 gene categories are built into the software. Define your own custom gene categories and use them for filtering.

Gene summary pages, Variation position summary pages with HGVS notation, Structural variation detail page, Copy number variation detail page, Genome reports, Keep notes on variations and genes, Organize genomes and create genome sets, Save bookmarks, etc.
Hardware and software requirements to run Enlis Genome Research on your own computer:
General requirements:
While our software should run on almost any standard desktop or laptop purchased in the last several years, it will of course run faster with better computer hardware.
Three components that make the most difference are:
Hard drive: Faster the better, 7200rpm disk drive or SSD
Memory: At least 2GB, more the better
CPU: Recent Intel or AMD
Hard drive space:
The fully installed program requires 9GB of space (this includes the 3 sample genomes).

Operating system:
Windows: Windows XP, Vista, 7, 8, or 10
Mac OSX: 10.6, 10.7, 10.8, 10.9, or 10.10
Linux: GTK+ 2.18 or higher, GLib 2.22 or higher, Pango 1.14 or higher, X.Org 1.7 or higher, libstdc++ 4.3 or higher
Each install package contains a full-featured version of the Enlis Genome Research software.
Contents:
Enlis Genome Research
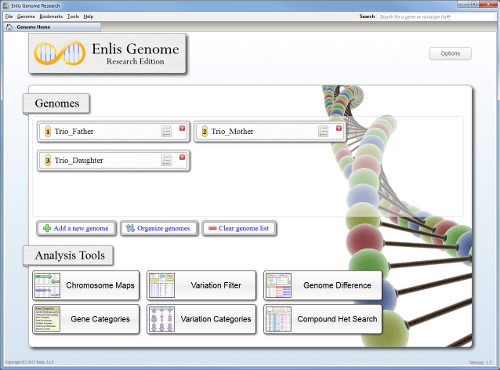
Main visualization and analysis software.
Enlis Genome Importer tool
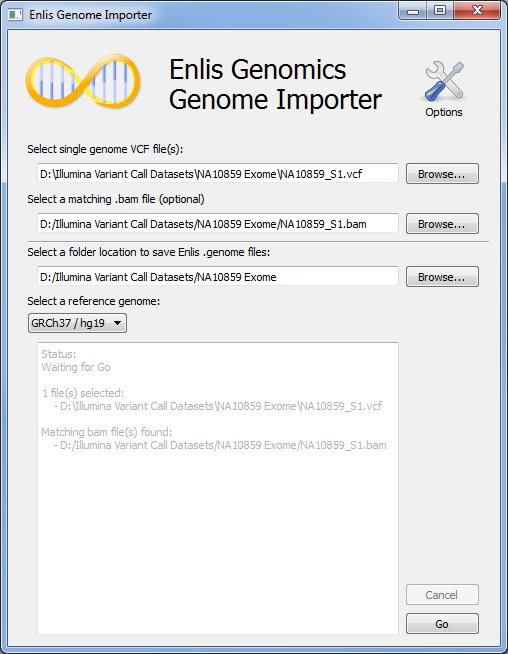
Point-and-click annotation to create Enlis .genome files.
Comprehensive annotation databases
No external downloads are necessary. All the required databases are curated by Enlis staff and included.
3 sample .genome files
Test out the software quickly by taking a look at the included family trio. (Whole genome data)
Pedigree 1463 Family Trio NA12877 (Father), NA12878 (Mother), and NA12882 (Son)
The sequences and variant calls were generated by Complete Genomics. Full annotation of the variations, call coverage, and metadata generation was done by Enlis with the genome importer tool.
Sign up today to request a free time-limited trial download:
Request free trial »